
2. How to bring your first small molecule, pharma product to the generic market
Part 2. Building Your Generic: How to Develop a Market-Ready Pharma Product
So, you’ve chosen your first generic drug product (see Part 1 if you haven’t yet). Now comes the exciting part—developing your version and preparing it for FDA submission. But what does “development” mean when you’re not the innovator?
In the world of generics, you’re not reinventing the molecule. You’re replicating a proven product—faithfully and efficiently—under tight regulatory constraints. The goal? To match the Reference Listed Drug (RLD) in performance, form, and safety… while building a dossier robust enough to satisfy the FDA.
Let’s walk through what that actually takes—from formulation to facility selection.
Defining “Development” for a Generic
For generic drugs, “development” doesn’t mean clinical discovery—it means:
Identifying the right formulation (including excipients)
Proving bioequivalence
Validating your manufacturing process
Documenting all of it for the FDA in an Abbreviated New Drug Application (ANDA)
But it all begins with a critical question: can you legally and scientifically replicate the RLD?
Know Your Constraints: What You Can (and Can’t) Change
There are four major areas where the FDA expects your generic to stay in line:
1. Ingredients
Stick to the RLD’s ingredients wherever possible—especially the active pharmaceutical ingredient (API).
Your API should come from a supplier with a Drug Master File (DMF) registered with the FDA. This lets you reference the supplier’s data rather than develop your own.
Even inactive ingredients (excipients) should match closely. Deviating may trigger additional justification, delays—or outright rejection.
2. Dosage Form
The generic must have the same dosage form, strength, and route of administration as the RLD.
You should also match the primary packaging, as this can affect stability and regulatory approval.
3. Critical Quality Attributes (CQAs)
These are the specific physical, chemical, and microbiological standards your product must meet.
Your CQAs should align with those of the RLD to prove safety, consistency, and therapeutic equivalence.
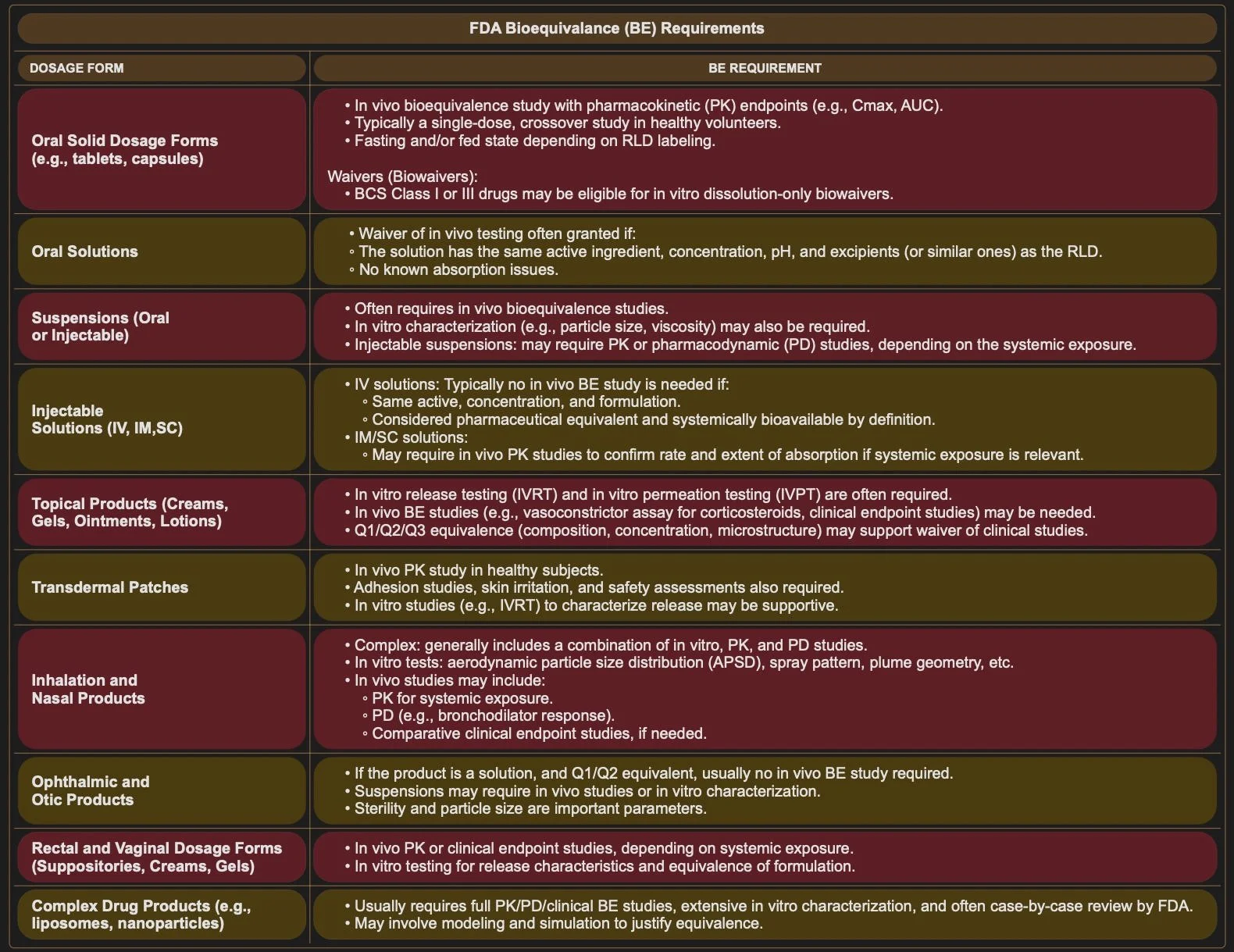
4. Bioequivalence (1)
You’ll need to demonstrate that your drug is absorbed at the same rate and extent as the RLD.
This is usually the most expensive part of generic development, and varies based on dosage form. Some dosage forms require a Bioequivalence (BE) study to be conducted on volunteer patients, In Vivo, whereas other dosage forms may qualify for a biowaiver and bioequivalence can be demonstrated by In Vitro data only.
If you are required to run a BE study, it can have a significant impact on the cost of your application and the tie it takes to prepares it.
Bioequivalence Studies: What Will It Cost?
Here’s a rough breakdown of typical BE study costs:

If your selected drug requires a BE study, factor this heavily into your ROI calculations..
Manufacturing: From Data to Drug
Once you’ve confirmed you can develop a compliant product, the next step is to find the right partner to manufacture it. Generic manufacturing comes in two flavours:
Development Manufacturing – Creating validation batches and generating data for your ANDA
Commercial Manufacturing – Producing the drug at scale once approved
Unless you’re running your own plant, you’ll need a CDMO (Contract Development and Manufacturing Organization). So how do you choose the right one?
Choosing a CDMO: What to Look For
Here are the five non-negotiables:
Regulatory Standing
Facility must be FDA-compliant and ideally recently inspected for a similar dosage form.
Must be GDUFA-compliant (Generic Drug User Fee Amendments).
Technical Capabilities
Can they handle your API and process without major changes?
Do they have the necessary analytical and processing equipment?
Capacity Fit
Is their batch size aligned with your projections?
A mismatch can hurt cost-efficiency and reliability.
No Conflicts of Interest
Are they already producing this drug for a competitor?
Will you get the attention and resources your project needs?
Location and Logistics
Is the facility easy to integrate into your supply chain?
Long-Term Considerations
Before you lock in your manufacturing partner, ask:
What’s your go-to-market strategy?
Will you sell the product under your brand?
Will you out-license it?
Will you register in other markets beyond the U.S.?
Each strategy has implications for your supply chain and documentation.
Can one facility do both development and commercial production?
Some CDMOs specialize in development but aren’t built for large-scale runs. Others can’t handle early-stage formulation work. Choose wisely—or be prepared to transfer the project midstream, which brings complexity and regulatory risk.
Next Steps: Turning Plans into Data
By this point, you should know:
What product you’re developing
How it compares to the RLD
Who will manufacture it
What constraints you’re working within
The final step? Generating the data for your ANDA.
Stay tuned for Part 3, where we dive into how to build the actual CTD—what studies are needed, how to document your process, and how to get your ANDA ready for FDA review.
Useful links: